Crystal Palace A program for Crystal Parametric Analysis, Least squares and Atomic Coordination with Estimated uncertainties
The organisation and analysis of crystal structure data from a parametric study (e.g. as a function of pressure and/or temperature) is a challenge!
Use of conventional programs that handle one refined crystal structure at a time is very time consuming and runs the risk of introducing
cut-and-paste errors into final tables. And a major shortcoming of crystallographic information format (cif) files as currently used is
that they do not contain the full variance-covariance matrix from the structure refinement, but only the uncertainties of the individual positional parameters.
Without the covariance of positional parameters, the esu’s of bond lengths and angles cannot be determined.
Crystal Palace is a new Windows program that solves these challenges for you:
It reads in your refined structure as a function of P or T or composition from a cif or cifs.
For information about cifs visit the IUCr website.
It identifies the equivalent atoms in each of the structures, even if they have different site names or occupancies.
It calculates bond lengths, angles, polyhedral volumes and distortion parameters, bond valences and thermal motion corrections to bond lengths.
It produces tables of all of these, ordered by pressure or temperature; no manual editing of tables ever again!
It uses symmetry to estimate the covariance of atomic coordinates and thus the esu's of structural parameters.
It can write new cifs and export unit-cell data and polyhedral volumes to the EosFit program to allow their thermal expansion and
compressibility to be fitted with equations of state.
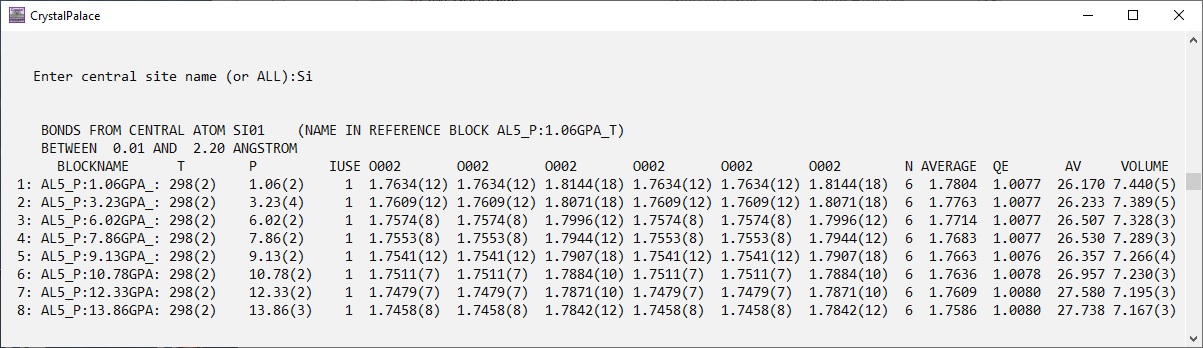
Here is an example of what Crystal Palace can do; the Si-O bond lengths in SiO2 stishovite as a function of pressure, calculated from the structures determined by Criniti et al (2022)
American Mineralogist 108, p 1559
Videos
Here are some short videos that introduce the basic features of Crystal Palace and show you how to use it.
To view the videos in full screen click on the 'picture-in-picture' icon
and then either drag the edges of the video window to make it larger or hit the full screen icon at the bottom right corner of the video window.
The one thing that Crystal Palace cannot do is draw pictures of crystal structures.
We use the CrystalMaker® software for all of our crystal structure drawings because
it can read the same cif files that we read into Crystal Palace. You can see some examples in the videos below.
1: The Introduction: view this one first!
We show you how to read crystal structure data into Crystal Palace from a cif, how to calculate bond lengths, angles and polyhedral volumes,
and how to save the results for using in plotting programs and writing papers.
Commands introduced:
Help
Read
List
Table
Calc
Log
Exit
2: Managing Crystal Structures
We show you how to combine multiple structures into a single cif, and how you can organise structures and output from Crystal Palace in different ways.
Commands introduced:
Clear
Sys and prog
Sort and unsort
Omit
Use
Export
Write
3: Understanding Polyhedra
In this video we discuss the concept of coordination polyhedra and the different parameters that Crystal Palace can calculate to describe polyhedral distortions.
4: A brief guide to thermal motion
Atoms in crystals are always moving and their motion is described in structure refinements by displacement parameters.
In this video we explain the basic concepts of displacement parameters and how to display them in Crystal Palace. We
discuss the limitations of conventional structure refinement results when the structure includes rigid bonds, such as in
tetrahedral frameworks like feldspars,
and show how to make thermal corrections to bond lengths.
5: An introduction to bond valence
The strength of an inter-atomic bond is quantified by its valence. In this video we introduce the concept of bond
valence and show you how to calculate bond valences in the Crystal Palace program.
Later videos will show you how to handle problems in bond valence calculations caused by problems in the cifs
and how to handle calculations in complex structures. So, if you get silly results from your bond valence calculation
after watching this video, look at the others!
6: Problems with your cif?
Sometimes the Crystal Palace program will not read the structure data from your cif. Or sometimes it will not
calculate the bond lengths and angles that you want. This is often caused by errors in the cif.
In this video we show you some common problems how to overcome them:
Start: data in data_global is not read in
9 mins: other cif errors
12 mins: warnings about symmetry
16 mins: no esu's calculated for bond lengths
20 mins: atoms not found; origin shifts and the Transform command
More videos will be added soon, so watch this space!
Crystal Palace Download and Installation
The Crystal Palace program is only available for Windows operating systems. Current version is 1 February-2025.
If you download and use Crystal Palace, please register with us by e-mail.
If you publish results from using the program, please support further developments by citing our paper:
Angel RJ, Mazzucchelli ML, Baratelli L, Schweinle CF, Balic-Žunic T, Gonzalez-Platas J, Alvaro M (2025)
Uncertainties of recalculated bond lengths, angles and polyhedral volumes as implemented in the
Crystal Palace program for parametric crystal structure analysis.Acta Crystallographica A, in press.
We recommend installing the program in a folder under C:\Program Files (x86).
You may have to authorise the installation of software 'from an unknown publisher'.
Installation may reboot your computer.
Installation includes example cifs and a copy of the manuscript describing the program. Copy these from the \examples folder of the installation
to a folder under \My Documents. The example cifs include some of those used in the videos.
Start the program from the short-cut on your desktop, and try to open one of the example cifs.
If you have problems, please consult the instructional videos first before contacting us.
If you download and use the Crystal Palace program, please register with us by e-mail.
This will ensure that you recieve information when program updates are released.
If you think you have found a bug in the software, please try and reproduce it with a simple example. Record a log file showing the problem,
and send it, your input cif, and a description of the problem to us at
rossangelsoftware@gmail.com
Comments and suggestions for improvements to the software, and requests for new features, are always welcomed.
Known bugs
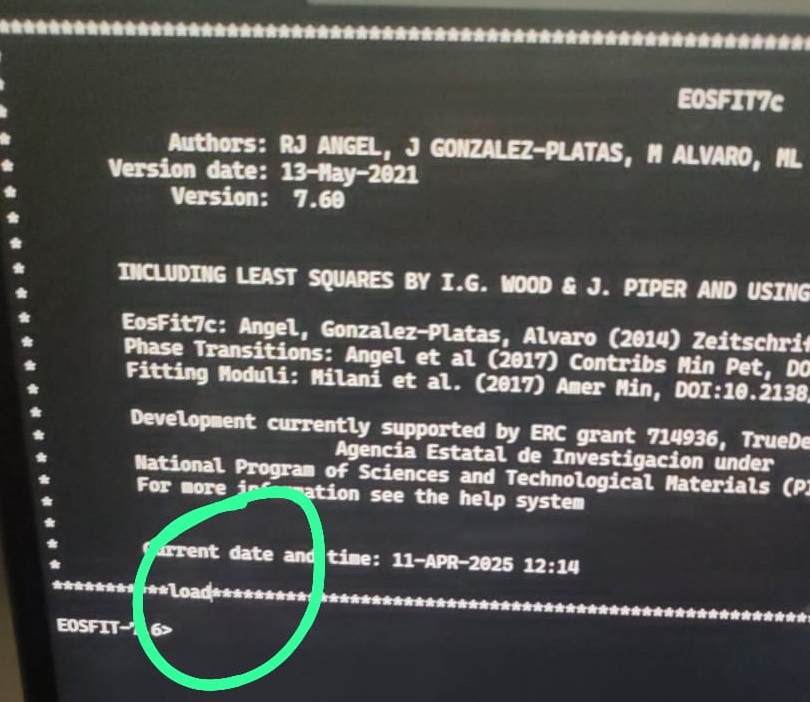
Windows11: If the commands you type in on the console appear above the prompt, like this in EosFit-7c:
then you need to adjust the size of the console emulator (why this is only a problem in Windows 11, nobody knows!):
Open Eosfit 7c
Right click on the top bar to bring up a menu
Click on Settings
Click on Launch Size in settings menu and increase or adjust (it depends on the screen resolution that you have)
the number of rows and columns: setting the rows to 50 and the columns to 140 seems to work on several computers.
Useful information sources
How to report estimated standard uncertainties: See recommendation 12 in
Statistical Descriptors in Crystallography. II. Report of a Working Group on Expression of Uncertainty in Measurement.
Acta Crystallographica A, 51, 565-569.
Polyhedral distortions defined by Robinson et al:
Quadratic Elongation: A Quantitative Measure of Distortion in Coordination Polyhedra.
Science, 172, 567-570.
Polyhedral distortion parameters defined by Tonci Balic-Žunic and co-workers:
Balic-Žunic, T. (2007) Use of three-dimensional parameters in the analysis of crystal structures under compression. In: Grzechnik A (Editor)
Pressure-Induced Phase Transitions. Transworld Research Network, Kerala, India. ISBN: 8178952726
Balic-Žunic, T., and Makovicky, E. (1996) Determination of the centroid or 'the best centre' of a coordination polyhedron.
Acta Crystallographica B, 52, 78-81.